YKSI ALATYYPPI JOKAISELLE ENTSYYMILLE
Kun elimistö sulattaa proteiinia se hajoaa aminohapoiksi. Jotkut näistä muuttuvat myrkyllisiseksi ammoniakiksi, joka muunnetaan maksassa myrkyttömäksi kemikaaliksi (urea). Tämä tehdään ureasyklinä tunnetulla prosessilla. Ureasyklissä tapahtuu useita vaiheita, joista jokainen vaatii entsyymin toimiakseen.
Vaikka UCD:t ovat geneettisesti erilaisia, ne jakavat tärkeitä ominaisuuksia ja siksi niitä käsitellään yleensä ryhmänä. Useimmille UCD-sairauksille on ominaista ammoniakin kertyminen vereen, jota kutsutaan “hyperammonemiaksi”.
On olemassa kuusi erilaista UCD-alatyyppiä – yksi kullekin ureasyklin entsyymille. Ureasyklisi alatyypin nimi edustaa entsyymiä, johon tämä vaikuttaa.
Jos sinulla on ureakiertohäiriö, olet syntynyt ilman jotakin kuudesta ammoniakkia hajottavasta entsyymistä tai tai sinulla on liian vähän sitä. Alta löydät eri UCD-alatyyppien nimet ja lyhyet kuvaukset.
Karbamoyylifosfaattisyntetaasi I -puutos (CPS1-puutos) on harvinainen geneettinen sairaus. Se johtuu Karbamoyylifosfaattisyntetaasientsyymin (CPS) vajauksesta tai puuttumisesta.
Ilman CPS-entsyymiä maksa ei voi muuttaa jäteproteiinia ureaksi yhtä nopeasti kuin normaalisti. Tämä voi johtaa korkeisiin ammoniakkipitoisuuksiin, erityisesti proteiinien lisääntyneen hajoamisen aikana.
CPS1-puutoksen oireet ja hallinta ovat samanlaisia kuin muiden UCD:iden.
CPS-puutos vaihtelee vakavuuden ja alkamisiän mukaan. Vastasyntyneen CPS:n oireet ilmaantuvat 24–72 tuntia syntymän jälkeen. Myöhemmin alkanut CPS1-puutos ilmaantuu yleensä lapsuudessa.
Lapset, joilla on CPS1-puutos, perivät yhden ei-toimivan CPS-geenin molemmilta vanhemmalta ja diagnoosi vahvistetaan löytämällä mutaatio CPS-geenistä.
Ornitiinitranskarbamylaasin puutos (OTC) on harvinainen geneettinen sairaus ja yleisin UCD-tyyppi. Se johtuu (OTC)-entsyymin vajauksesta tai puuttumisesta.
Ilman OTC-entsyymiä maksa ei pysty muuttamaan jäteproteiinia ureaksi yhtä nopeasti kuin normaalisti. Tämä johtaa korkeisiin ammoniakkipitoisuuksiin, erityisesti proteiinien lisääntyneen hajoamisen aikana.
OTC-puutoksen oireet ja hoito ovat samanlaisia kuin muut UCD:t.
OTC-puutos vaihtelee vakavuuden ja iän mukaan. Se on X-kytketty sairaus, mikä tarkoittaa, että OTC-geeni löytyy X-kromosomista. Naisilla on kaksi X-kromosomia, kun taas miehillä on yksi X-kromosomi ja yksi Y-kromosomi. Koska miehillä on vain yksi kopio geenejä X-kromosomista, kaikilla miehillä joilla on OTC-puutos on ongelmia. Naisilla, joilla on OTC-geenin mutaatio ei ehkä koskaan ole ongelmia, koska heidän toinen kopionsa geenistä (toisessa X-kromosomissa) on normaali – heitä kutsutaan kantajiksi. Miehillä esiintyy yleensä OTC-puutosoireita 24–72 tuntia syntymän jälkeen, mutta vain noin 25 prosentilla naisista ilmenee oireita lapsuudessa.
Diagnoosi vahvistetaan löytämällä mutaatio OTC-geenistä.
arginiinimeripihkahappolyaasin puutos (ASL), on harvinainen geneettinen sairaus. Se johtuu arginiinimeripihkahappolyaasientsyymin (ASL) vajauksesta tai puuttumisesta.
Ilman ASL-entsyymiä maksa ei pysty muuttamaan jäteproteiinia ureaksi yhtä nopeasti kuin normaalisti. Tämä voi johtaa korkeisiin ammoniakkipitoisuuksiin, erityisesti proteiinien lisääntyneen hajoamisen aikana.
ASL-puutoksen oireet ja hoito ovat samanlaisia kuin muiden UCD-tautien.
ASL-puutos vaihtelee vakavuuden ja iän mukaan. ASL-potilailla voi esiintyä oireita pian syntymän jälkeen vaikeassa muodossa (24–48 tunnin kuluessa) tai lapsuudessa ASL-entsyymin osittaisesta puutteesta.
Lapset, joilla on ASL-puutos, perivät yhden toimimattoman ASL-geenin jokaiselta vanhemmalta ja diagnoosi vahvistetaan löytämällä mutaatio ASL-geenistä.
Arginiinimeripihkahapposyntetaasin puutos (ASS), jota kutsutaan myös tyypin 1 sitrullinemiaksi (CTLN1), on harvinainen geneettinen sairaus. Se johtuu arginiinimeripihkahapposyntetaasin (ASS) puutteesta.
Ilman ASS-entsyymiä maksa ei pysty muuttamaan jäteproteiinia ureaksi yhtä nopeasti kuin normaalisti. Tämä voi johtaa korkeisiin ammoniakkipitoisuuksiin, erityisesti proteiinien lisääntyneen hajoamisen aikana.
Erittäin korkeiden sitrulliinitasojen kertyminen erottaa ASS-puutoksen muista UCD:n alatyypeistä.
ASS-puutoksen oireet ja hoito ovat samanlaisia kuin muilla UCD:n alatyypeillä. ASS-potilailla voi esiintyä oireita 24–72 tunnin sisällä syntymästä tai lapsuudessa.
Lapset, joilla on ASS-puutos, perivät yhden toimimattoman ASS-geenin jokaiselta vanhemmalta. ASS-geenissä on ohjeet ASS-entsyymin tekemiseen ja diagnoosi vahvistetaan löytämällä mutaatio ASS-geenistä.
Arginaasi 1:n puutos (ARG1-D), jota kutsutaan myös hyperargininemiaksi, on harvinainen geneettinen sairaus. Se johtuu arginaasi 1 (ARG1) -entsyymin vajauksesta tai puuttumisesta.
Ihmiset, joilla on ARG 1 -puutos, voivat ilmaista oireita myöhemmin kuin ihmiset, joilla on muita UCD-tautia, eikä hyperammonemia ole yhtä vallitseva tai vakava.
Sairastuneilla lapsilla saattaa esiintyä jäykkyyttä (spastisuutta), oppimisvaikeuksia, kouristuksia ja heikkoa kasvua veren korkeiden arginiini- ja ammoniakkipitoisuuksien seurauksena. Yleensä lapsilla, joilla on ARG 1 -puutos, alkaa kehittyä kävelyhäiriöitä ja spastisuutta 2-3 vuoden iässä.
Lapset, joilla on Arginaasi 1 -puutos, perivät yhden toimimattoman ARG1-geenin jokaiselta vanhemmalta. Plasman arginiinin korkeiden tasojen kertyminen erottaa ARG1-D:n muista UCD:istä ja diagnoosi voidaan vahvistaa löytämällä mutaatio ARG1-geenistä ja korkeat arginiinitasot verestä.
N-asetyyliglutamaattisyntetaasin puutos (NAGS) on harvinainen geneettinen sairaus. Se johtuu N-asetyyliglutamaattisyntetaasin (NAGS) puutteesta.
Ilman NAGS-entsyymiä maksa ei pysty muuttamaan jäteproteiinia ureaksi yhtä nopeasti kuin normaalisti. Tämä voi johtaa korkeisiin ammoniakkipitoisuuksiin, erityisesti proteiinien lisääntyneen hajoamisen aikana.
NAGS-puutoksen oireet ja hoito ovat samanlaisia kuin muut UCD:t. Ihmiset joilla on NAGS voivat oirehtia 24–72 tunnin kuluessa syntymästä tai myöhemmällä iällä.
Lapset joilla on NAGS-puutos, perivät yhden toimimattoman NAGS-geenin jokaiselta vanhemmalta. NAGS-geenillä on ohjeet NAGS-entsyymin valmistamiseksi. Diagnoosi vahvistetaan löytämällä mutaatio NAGS-geenistä.
Lisäksi on olemassa kaksi entsyymien kuljettajan puutetta, joita pidetään myös UCD:inä:
Ornitiinitranslokaasin puutoksesta johtuva hyperornitinemia-hyperammonemia-homositrullinuria-oireyhtymä (HHH-oireyhtymä).
Sitriinin (tyypin 2 sitrullinemia) puutos
UREAKIERTO
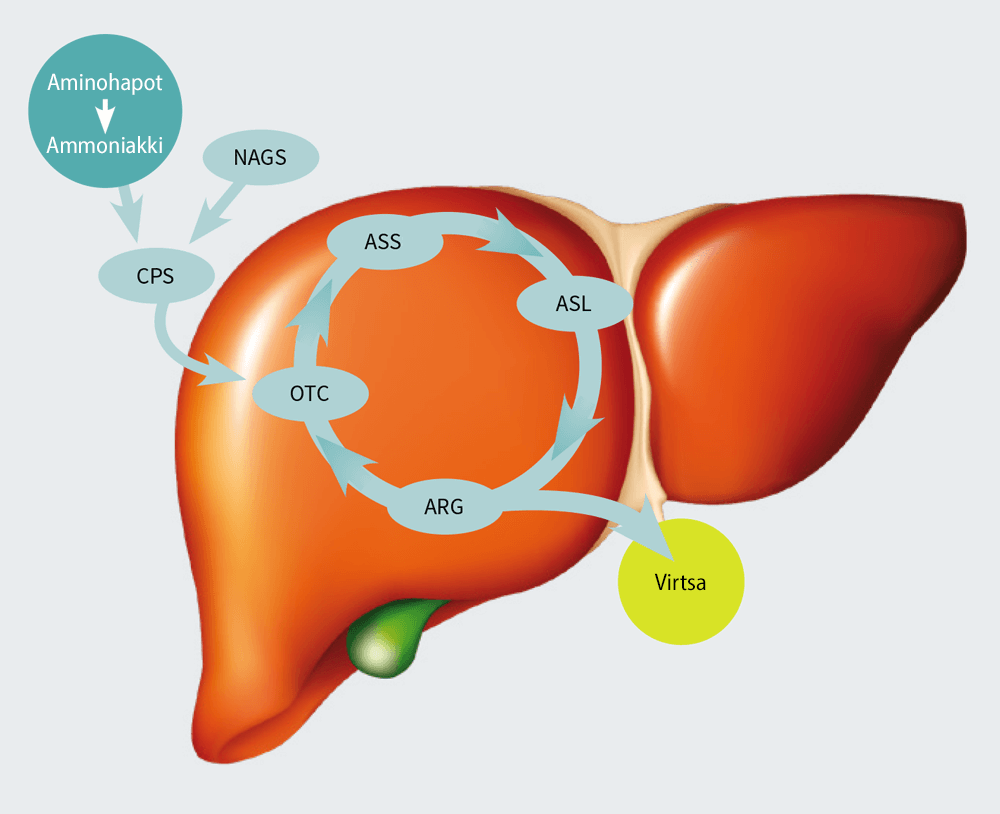
Ureakierto tapahtuu maksassa. Kunkin UCD:n nimet edustavat entsyymiä, johon ureakierto vaikuttaa. Maksan entsyymit muuttavat ammoniakin ureaksi, jotta elimistö pääsee eroon siitä virtsan kautta.